

Obesity's relationship with type 2 diabetes is really weird

Obesity is a really complicated subject. I once had a 3 month long project trying to develop a therapeutic for obesity-related asthma stymied by the graph below.

I was completely unable to explain why obesity-related asthma (which is generally more serious and more treatment-resistant than normal asthma) only seemed to affect obese women, and so I had to give up my proposed therapeutic and turn to autoimmune diseases instead.
That being said, obesity is also a fascinating and important subject, which is why I keep returning to it. Obesity has become a global epidemic, spreading almost like a disease. It’s almost always associated with worse health outcomes in a host of different conditions (at least 13, by the CDC’s count). And, despite our best efforts, we still don’t have a single good answer as to the cause of obesity’s rapid rise in prevalence.
In this essay, I want to zoom in on a really specific disease that’s heavily associated with obesity: type 2 diabetes. Type 2 diabetes, if you don’t remember, is the type of diabetes that’s associated with insulin resistance, rather than insulin-producing cell destruction (and if you want a brief refresher on insulin and its relation with glucose/blood sugar in general, follow the footnote [1]).
Now, obesity causes diabetes. That much is indisputable. Compared with adults with normal weight, adults with a BMI of 40 or higher had an odds ratio (OR) of 7.37 for diagnosed diabetes, which is comparable to the odds ratio of ever smoking for lung cancer. Furthermore, we can see that there’s a direct correlation of weight (and more specifically, waist circumference) with insulin resistance, which leads directly to type 2 diabetes. Basically, if you’re obese, type 2 diabetes is a really serious health risk, and one of the main causes for early mortality of obese people.
When we look into why this happens, however, things get a lot murkier. Part of the problem is that a lot of things happen when a person becomes obese like growth of fat cells, an increase in body-wide inflammation, and a change in the microbiome. You could conceivably link all of these to insulin resistance (and people have), although none of them have an easy mechanical link in the same way as, say, obesity and high blood pressure.
Instead of pursuing difficult-to-falsify correlative chains, though, let’s approach the same problem from a different angle. Instead of asking why obesity causes type 2 diabetes, let’s ask how we could reverse type 2 diabetes, and see if that gives us any ideas about the cause. Once we do so, we’ll see that things get really weird.
So, weight gain causes insulin resistance which causes type 2 diabetes. Naively then, we might think the best way to reverse type 2 diabetes would be weight loss.
And, funnily enough, that actually works. Every successful method of weight loss that I’m aware of results in at least a temporary drop in insulin resistance and a temporary cessation of the need for type 2 diabetics to use insulin. This includes bariatric surgery, fasting, eating at a caloric deficit from a low-carb or low calorie diet, and the GLP-agonist semaglutide.
Here’s the weird part, though. For every one of those methods that there’s data for, the drop in insulin resistance occurs before the vast majority of the weight loss. For example, the below graphs are from a paper showing the effects of Roux-en-Y Gastric Bypass surgery on obese diabetics and non diabetics.
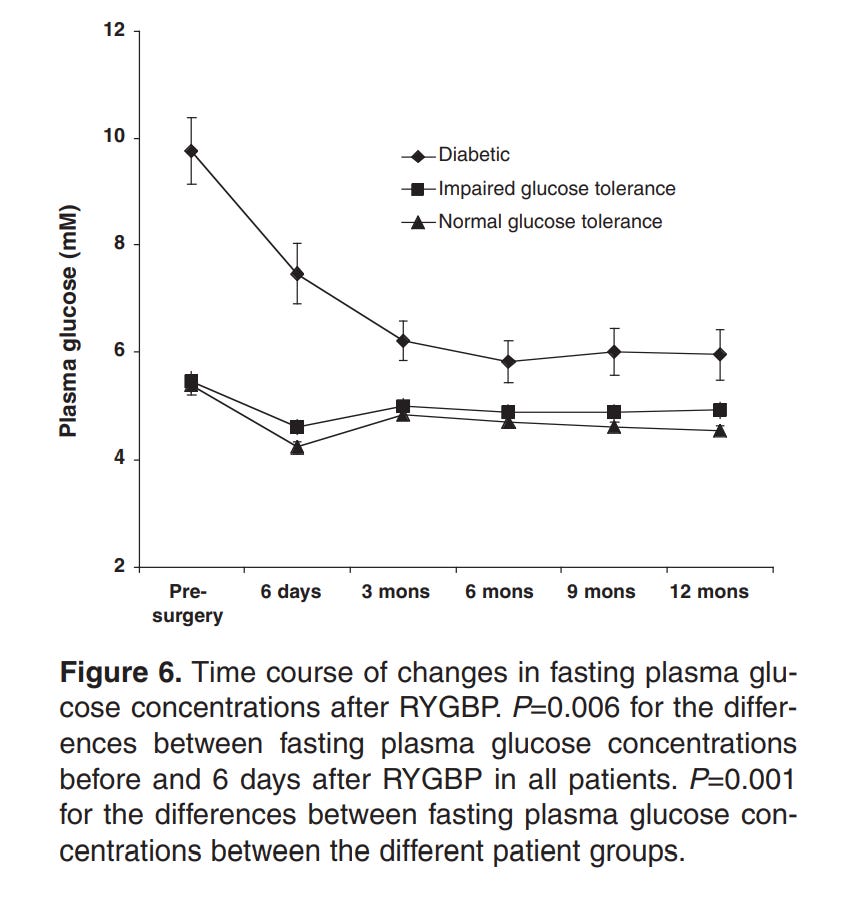
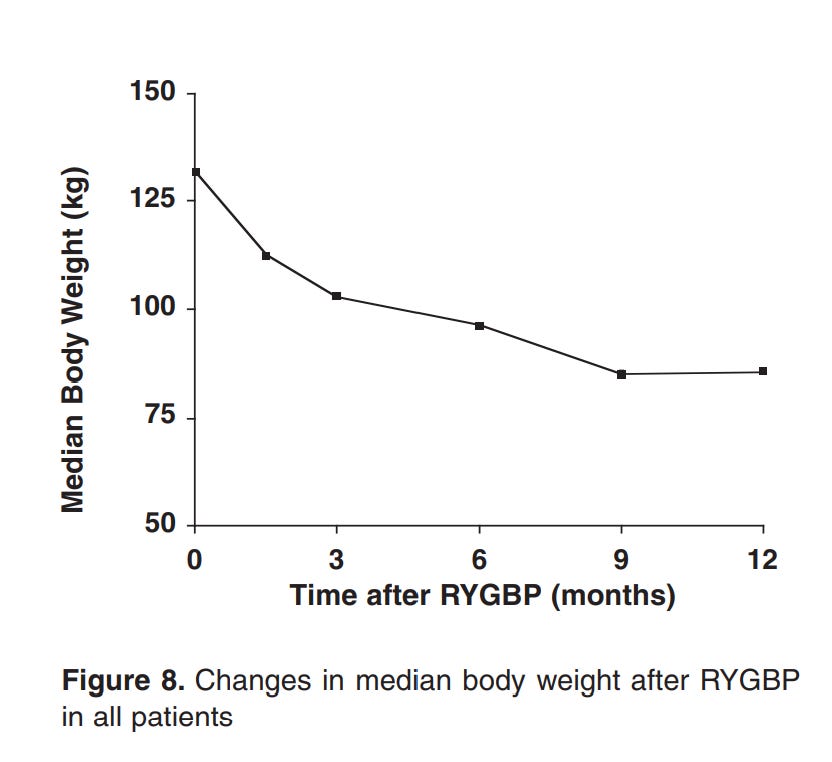
Note how there’s a 20% loss in glucose in a period of days, while the equivalent weight loss only occurs 3 months in.
The same pattern holds with fasting and with caloric deficits. The loss in insulin resistance comes before the weight loss. To make things even stranger, for fasting and caloric-deficit diets, the insulin resistance bounces right back as soon as the subjects resume their normal diets. Presumably, the same results would hold true for gastric bypass surgery too, but it’s not really possible to easily resume a normal diet after gastric bypass.
So, the next thing we might think is, “What’s a process that occurs quickly after fasting and stops after resuming a normal diet?” The answer to this would obviously be lipolysis, the breakdown of fat cells into fatty acids for the purpose of providing energy. The other side of this would then be that lipogenesis, the creation of fat cells, would be the cause of insulin resistance. In other words, it’s not being obese that causes insulin resistance, but becoming obese.
And, lo and behold, we get some evidence towards that, too. Namely, there’s an observed strong correlation between lipogenesis in the liver and insulin resistance. In the past (and in the linked paper), it’s been assumed that insulin resistance causes lipogenesis, but the evidence could easily be used to support the opposite.
This is all perfect. Mystery solved. Or at least, it would be, if obesity wasn’t so damn complicated.
Because, unfortunately, you know what else causes a drop in insulin resistance, both in mice and humans? Inhibiting lipolysis, either genetically or pharmacologically. Yup, literally the opposite of what I just suggested. Inhibiting lipolysis also causes an uptick in lipogenesis in some parts of the body (although a decrease in others), leading us to the unfortunate observed correlation that, at least in this case, lipogenesis is also correlated with a drop in insulin resistance.
To be frank, that’s where I’m stuck. I don’t have a great explanation for this without venturing into some less-supported territory. However, if you are willing to clamber out with me on some metaphorical thin ice, then let’s proceed.
Let’s go back to our odd guy out in weight-loss strategies: the GLP-1 agonist, semaglutide. GLP-1 stands for glucagon-like peptide 1, a hormone that comes from the same precursor as glucagon, proglucagon (glucagon, if you don’t remember, is the opposite of insulin, in that it promotes the creation of blood sugar). GLP-1 is released from the intestines shortly after a meal to promote the secretion of insulin and so the uptake of glucose from the bloodstream.
What makes semaglutide and other GLP-1 agonists interesting is that they promote the secretion of insulin while promoting weight loss. This is really surprising, as direct administration of insulin causes weight gain, which is one of the cruel ironies of type 2 diabetics needing to take insulin. GLP-1 agonists seem to do this by simultaneously inhibiting both lipolysis (through the secretion of insulin) and hepatic lipogenesis, which suggests that the two don’t have to be mutually exclusive (i.e. you can have neither net lipolysis nor net lipogenesis).
This would also explain why inhibition of lipolysis in the mouse study above didn’t result in the net creation of fat mass in the mice. If lipolysis is inhibited but there’s no net lipogenesis (even if there is specific lipogenesis), there will still be an excess of fatty acids in the bloodstream. This signals to your body that there’s no need for any more glucose in your bloodstream right now, as there’s enough energy in the form of fatty acids. This would also result in a decrease in appetite, as normally a decrease in blood sugar would prompt an increase in appetite.
Tying it all together, it’s all about the level of fatty acids in the bloodstream. If it’s high, whether because of active lipolysis or no net lipogenesis, insulin resistance decreases. If it’s low because of net lipogenesis, insulin resistance increases. We can also argue this is because low fatty acids allow for spillover of fatty acids into the bloodstream.
Personally, I like this theory, and I think it’s great. The real problem with it is that the data to back it up isn’t really there. Or, more to the point, there’s too much data, and it all points in different directions. I can cite studies that support my hypothesis and say high levels of FFA are inversely related to insulin resistance or I can cite studies that go against my hypothesis and say high levels of FFA are associated with insulin resistance.
Frankly, it feels like a measurement problem. Measuring hydrophobic molecules with short half-lives is hard, and it’s tough to know who’s doing it correctly, especially when you’re looking at old papers. Until that’s settled, there’s only one thing to do, and assume my theory is 100% correct.
[1] Glucose, as you likely know, is your body’s standard way of storing and transporting energy. If you have no glucose, you die. If you have too much glucose, you’re usually ok temporarily, but chronically high levels of glucose causes weird warping of organs and body systems that aren’t meant to handle those high levels, and you end up with bad effects like nerve damage and blindness.
So, your body tries to keep glucose levels in a healthy range. This is complicated by the fact that not only do you use glucose as fuel, but you also randomly take in large amounts of it whenever you eat a meal. So, your glucose (or blood sugar) levels end up looking like this throughout the day.

In order for your glucose levels to remain within a range, your body has a couple hormones it uses as signals for your cells to either take up glucose or to increase the production of glucose. Insulin is the main signal for your cells to take up glucose. As you might imagine, this sort of bodywide signal has a lot of downstream effects, as the act of taking up glucose prompts and causes many other processes to occur.
So far, so good. Insulin resistance is what it sounds like: your body cells don’t react to the insulin signal in the same way they used to. A downstream result of this is that blood glucose levels remain high despite the production of a certain level of insulin, prompting the body to produce even more insulin. If insulin resistance escalates, at some point the pancreas can’t produce enough insulin, which is when type 2 diabetes develops (there’s also some complicated relationship with the pancreatic cells working overtime and then actually losing the ability to produce insulin, but let’s leave that for now).
Type 2 diabetes, then, is defined by an excess of blood glucose caused by not enough insulin. There’s are a few different ways of diagnosing it: excessively high blood glucose when fasting, excessively high blood glucose after eating glucose, a high percentage of hemoglobin chemically bonded to glucose, or, my personal favorite, the old school method of tasting urine and seeing if there’s glucose in it (in a normal person, the kidneys filter out all the glucose from the urine before it’s eliminated).
I think coming back to the view of insulin resistance as an early symptom (preceded by hyperinsulineme but with normal glucose levels) of energy excess ties this all together. All this is from "from Obesity to Type 2 diabetes", details are probably wrong since its been a few yrs since I read it. This also ignores the role of the liver.
Basic logic is:
1. all humans (excepting those w/ rare diseases of fat cell differentiation) have a capacity for enegy buffering/storage because this is adaptive and a generally well-preserved trait in other animals.
2. in times of caloric excess, we increase fat deposition to store excess calories. This is mediated substantially through insulin. Biochemically, you'd see this with rising levels of insulin hormone production, with preserved/normal blood sugar levels.
3. Different people have differing capacities for insulin hormone production and different levels of fatness at which their capacity for energy storage becomes more resistant.
4. At this point, glucose levels start to rise, as insulin resistance rises and insulin hormone levels approach their max. This is prediabetes. At this point, these higher blood glucose levels are neccesary to keep driving calories into fat cells, even as those fat cells start to signal their full through insulin resistance.
5. At some point, your insulin levels can't rise further, and glucose levels reach "diabetic levels".
6. You have several therapeutic options.
A. Raise how much fat you can carry (thiazolidinediones do this)
B. Increase insulin production (exogenous insulin, sulfonyureas)
C. Reduce sugar intake (acarbose)
D. Reduce calorie intake (weight loss, GLP-1 agonists)
E. Reduce hepatic glucose output (Metformin)
From that perspective, it is easy easy to see why weight loss can so rapidly normalize symptoms. It is treating the fundamental underlying driver. Even mild weight loss can quickly move your equilibrium back to within-buffering range.
Further caveats: ignores pancreas burnout, ignores other hormonal effects of bariatric surgery and GLP-1 agonists, ignores hepatic glucose output, etc.
D. Increase sugar waste (SGLT-2-inhibitors do this, "safety valve" for high sugar levels)
Type of fat... I do believe if you consider the type of fat (trans, saturated vs. MUFA/PUFA), the lipolysis/lipogenesis confusion is less confusing, as inflammation feom lipotoxicity is actively being studied as a cause of insulin resistance.